随着耐甲氧西林金黄色葡萄球菌以及耐万古霉素粪肠球菌等多重耐药致命病原微生物的不断加重,2014年,世界卫生组织宣布人类进入后抗生素时代。换言之,从这个世纪开始,人类可能死于普通的感染或者小型创伤。同时,近30年来没有主要的新型抗生素被发现,人类开发新型抗生素迫在眉睫。2015年,Lewis等人发现了一种新型抗生素并命名为Teixobactin。其杀死小鼠机体中致命耐药细菌的速度与现存抗生素的速度相当,而且无毒副作用,安全有效。最关键的是其并不会诱发细菌耐药性的产生,这一发现也是近30年来的首次。其作用靶点是构成细胞壁的前体lipid II和lipid III,由于它们结构保守所以不易产生耐药性。Teixobactin 被称为“游戏规则颠覆者”,并且得到了世界范围内广泛的关注。受限于复杂的非天然氨基酸Enduracidine,到目前还没有实现天然产物teixobactin的放大量全合成。Teixobactin与靶点的具体的结合模式也没有得到很好的探究。
2019年7月22日,清华大学药学院饶燏课题组在国际学术期刊《自然·通讯》(Nature Communications)在线发表了最新研究成果《Teixobactin的克级全合成推动的其结合模式探索以及开发更强抗生素的研究》(Gram-scale Total Synthesis of Teixobactin Promoting Binding Mode Study and Discovery of More Potent Antibiotics)。
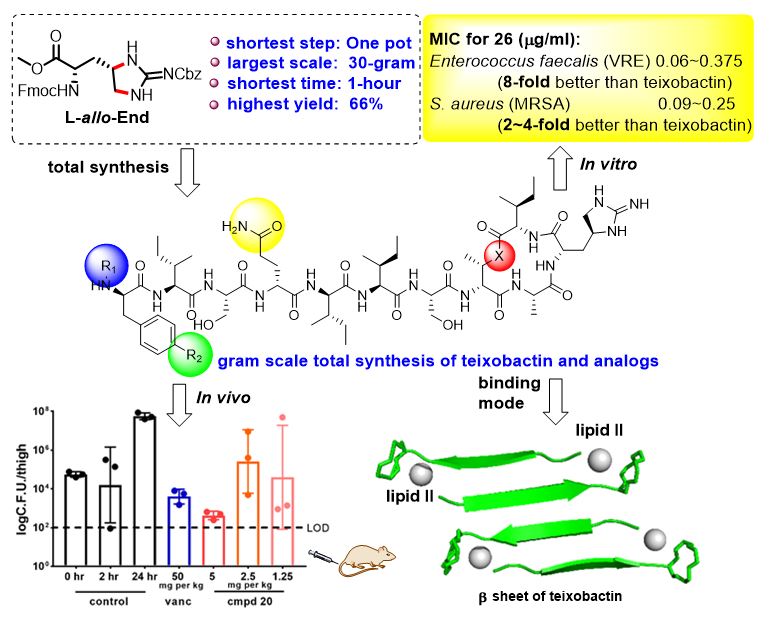
结构复杂、合成繁琐的非天然氨基酸Enduracidine的存在妨碍了科学家对于teixobactin的合成与研究,作者首先通过发展新的合成策略,从便宜易得的原料出发,实现了一锅法大量制备非天然氨基酸Enduracidine。此方法具备规模大、步骤少、耗时短、选择性好以及产率高等优点,反应总耗时1小时,规模至少30克。这也为接下来该课题组实现teixobactin的克级全合成打下了坚实的基础。
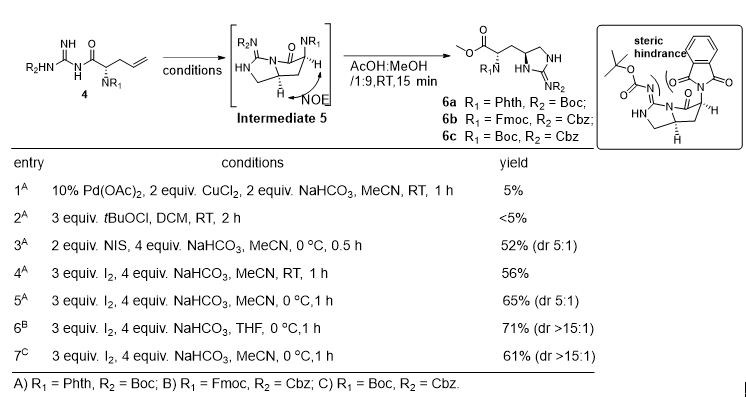
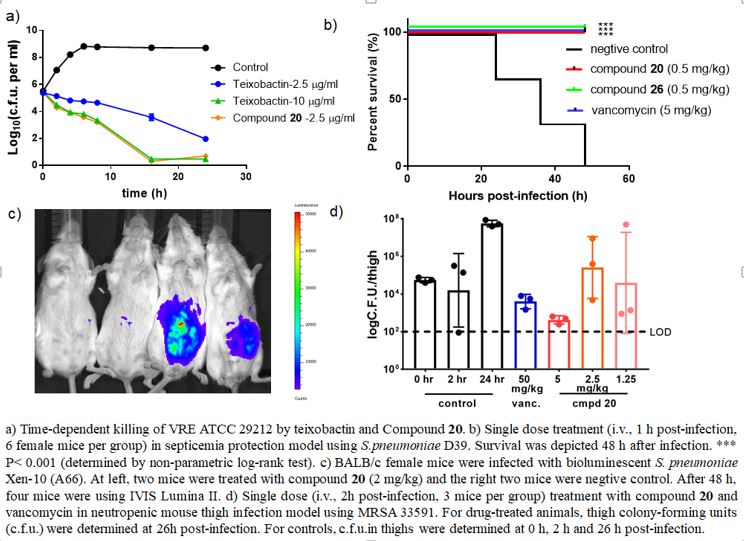
接下来,该课题组通过3+2+6的汇聚式合成策略,成功避免了前人在全合成中的副反应,首次实现了teixobactin的克级全合成。由于采取汇聚式合成路线,在构效关系研究的过程中,只需要对相应的合成模块进行改造,即可快速得到一系列基于teixobactin的抗菌库。最终该课题组发现类似物20和26展现出了更强的抗菌活性,这两个候选化合物近一步在动物体内展现了强大的抗菌活性。
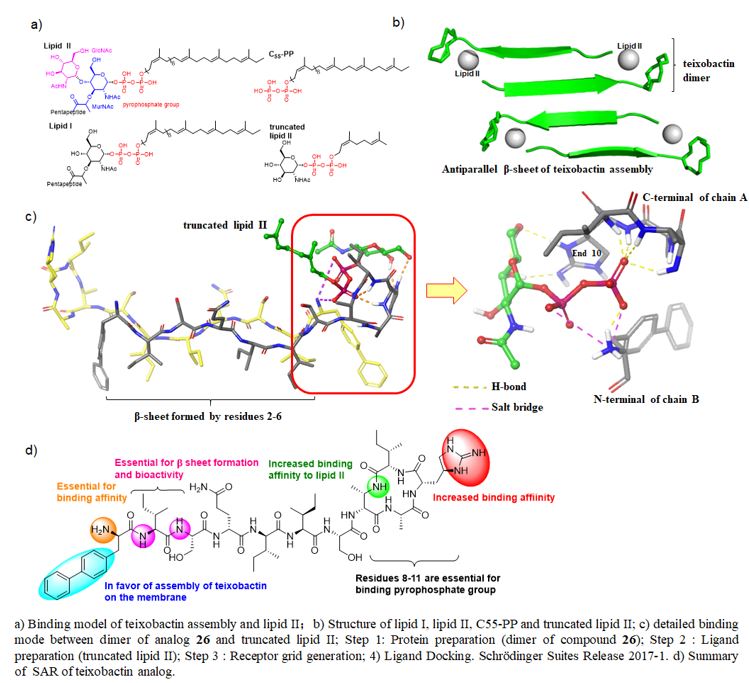
作者最后通过改变氨基酸,破坏相邻两条链的氢键相互作用,提出反相平行teixobactin形成的多聚体是其发挥抗菌活性的前提;通过分子对接,作者推测teixobactin反相平行的二聚体是结合靶点lipid II的最小结合单元,并且提出了可能存在的具体分子结合模式。
饶燏课题组一直致力于基于teixobactin开发新型抗生素。2018年在药物化学权威杂志Journal of Medicinal Chemistry报道了基于teixobactin开发新型抗生素类似物的研究工作。
这是一个较系统的药物化学基础研究工作,从发展新合成策略实现关键非天然氨基酸Enduracidine的扩大量合成,到实现天然产物teixobactin克级合成,再到基于teixobactin开发更强抗生素,最后近一步阐述其发挥抗菌活性的分子机制。该工作为基于teixobactin进行抗多重耐药菌的药物研发提供了重要信息,目前该团队研究人员在进一步进行此类新型化合物的开发工作。该工作由清华大学药学院饶燏课题组完成。药学院博士研究生宗昱,研究生方芳为共同一作,饶燏教授为本文通讯作者。该研究得到了完美体育·(中国)官方网站张敬仁教授以及美国东北大学Kim Lewis教授的支持和帮助,得到了清华大学生命科学学院薛毅教授的帮助,该研究得到了国家自然科学基金以及国家重大新药创制项目的资助,同时也得到了教育部蛋白质重点实验室和有机磷重点实验室的大力支持。
全文链接:https://www.nature.com/articles/s41467-019-11211-y













 学院动态
学院动态
